Appendix B.1 - Text-based User Interface (TUI) to PAMoC
The PAMoC text-based user interface (TUI) drives the user through a sequence of menus, each of which is displayed individually on the screen. In each menu the user must choose the most appropriate option, which in turn can request additional input.
The user can access the PAMoC-TUI by launching the executable code from his preferred Unix/Linux shell interpreter (e.g. sh, ksh, csh, tcsh, bash):
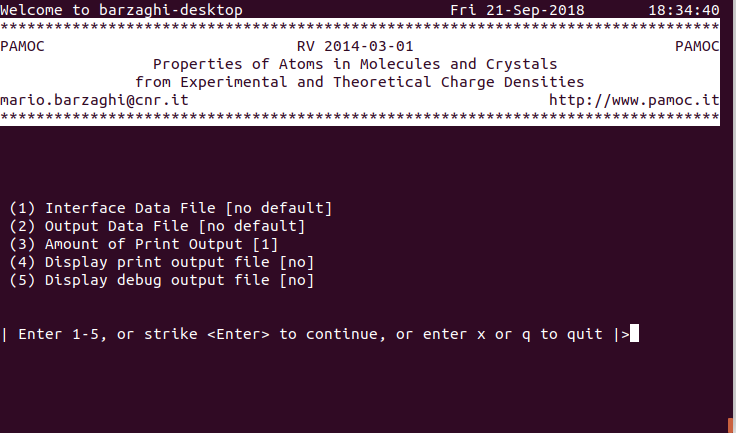
PAMoC-TUI is a separate version of PAMoC that uses an interactive text-based user interface and shares the rest of the code with PAMoC. Before entering into the interactive procedure, PAMoC-TUI performs all the calculations it can do, based on the input data acquired through the command-line keywords and options, and possibly the environment variables and the configuration file. In this section, we describe only the initial menu, which pops up as soon as the PAMoC-TUI is launched. Figure 1 shows the menu for the case where no input data of any type are given. Selecting option 1, you can enter the pathname of an interface data file (IDF), but you may not need an IDF. Option 3 is effective only if the pathname of an output data file is provided by selecting option 2. Similarly, setting option 4 to "yes" is effective only if an output data file has been assigned to the current job. Finally, setting option 5 to "yes" is effective only if the amount of print output (option 3) is set to 9 or higher, because in this case a debug-file (fort.99) is generated.
Please note that setting options 4 and 5 to "yes" is equivalent to opening two new terminals yourself, and giving the following command at the prompt:
If, in front of the menu in Figure 1, you decide to leave the PAMoC-TUI,
by entering one of the characters 'x', 'X', 'q', 'Q' at the terminal prompt,
you will see the same printout that is shown in the Tutorial 0Tutorial 0: launching
PAMoC without input data.
(click to display the printout in a
separate tab), scroll away on the screen. Of course, if you had
remembered to define an output file, the printout would have been sent to
that file. It is strongly suggested to always define the output file in order
to ensure a permanent historical record of all the actions taken and the
results obtained by PAMoC during the interactive session.
Interface Data File (IDF): /home/barzaghi/Work/gamess/h2o-2dfp.wfn Configuration File (CFG): pamoc.rc Output Data File (OUT): pamoc.out Debug Output File: fort.99 Options: IPrint 9 Debug T Interface (stop after) F Moments (stop after) F ECryst (stop after) F NoECryst F TopologyOnly F UseTopo2 F Grid F ChkTopo2F F UseSymmetry F Crystal F ChargeNorm F Bohr F NoVariance F DiagVCMatrix F DbgNuMom F DFT F Export DMA F NoMolRec F ScalePop F NoElProp F NoCrystalTop F Include Loose Hydrogen Bonds F READ TOPology from disk file F SAVE TOPology on disk file F QTAIM partitioning F EP/MM F E_Cryst extended Printing F lmx 4 Integration Grid Selection 11 Moment Origin 0 UseDMA 0 CluDim 1 MaxErr 3 Atoms to be integrated (QTAIM) 0 Dimer 0 E_Cryst Model 2 Atomic Repulsion Parameters 5 Atomic Dispersion Parameters 5 Damping Function for Dispersion 4 Modify Density Basis Set 0 EPMM Penetration Threshold J/mol 500 Moment shift procedure (0|1|2|3) 2 Molecular moments printing optns 2213 Radial quadrature rule 31 Standardization of Radial Grid 0 Order of Knowles Grid 3 Order of Handy Grid 2 Definition of covalent radii 0 DMA printing options 10011 Environment Variables: USER barzaghi PWD /home/barzaghi/src/pamoc/newp1 TERM xterm-256color DISPLAY :0 The first word on file: /home/barzaghi/Work/gamess/h2o-2dfp.wfn is Wate ******************************************************************************* PAMOC RV 2014-03-01 PAMOC Initialization of Data from Input- and Interface-data files pamoc.rc /home/barzaghi/Work/gamess/h2o-2dfp.wfn mario.barzaghi@cnr.it https://www.pamoc.it ******************************************************************************* Type of Interface Data File (IDF) ... formatted (ascii) ------------------------------------------------------------------------------- Contents of the wavefunction file ------------------------------------------------------------------------------- File name ............ /home/barzaghi/Work/gamess/h2o-2dfp.wfn Nr. of wavefunctions = 1 Title ................ Water, 6-31G(2df,p) basis Wavefunction type .... RHF SCF Total Density Nr. of electrons ... = 10 Nr. of MOs ......... = 5 Nr. of Primitives .. = 64 Nr. of Nuclei ...... = 3 ------------------------------------------------------------------------------- Dynamic thresholding algorithm to enable neglecting of small primitives (J. Cioslowski, Chem. Phys. Lett. 1992, 194, 73-78) NMO = 5 NPrims = 64 Cutoff parameter = 4 ( 0.55902E-05) Processing of the Interface Data File ... done Cell constants, Symmetry operations, etc. ... done Atom sites in the crystal/molecule ... done Symmetry ... done Density Basis Set ... done Mulliken and Stone DMAs ... done Evaluation of Atomic Charges by the Electronegativity Equalization Method ------------------------------------------------------------------------- EEM parameters A and B with kappa = 0.44, and estimated atomic charges ------------------------------------------------------------------------- I Element A B Atomic Charge ------------------------------------------------------------------------- 1 8 O 2.625 0.858 -0.410 2 1 H 2.396 0.959 0.205 3 1 H 2.396 0.959 0.205 ------------------------------------------------------------------------- Electronegativity Equalization Method: Parameterization and Validation for Large Sets of Organic, Organohalogene and Organometal Molecule Radka Svobodova Varekova, Zuzana Jirouskova, Jakub Vanek, Simon Suchomel, and Jaroslav Koca, Int. J. Mol. Sci. 2007, 8, 572-582. EEM populations ... done Nuclear Centred Multipole Moments ... done Evaluation of molecular descriptors ... done Initialization Step cpu time: 0.0 seconds. Common /DMAStat/ IStatDMA(0:MxDMA) : 1 111 111 0 0 0 0 0 0 111 default dma 1 dma 1 T dma 2 T dma 3 F dma 4 F dma 5 F dma 6 F dma 7 F dma 8 F dma 9 T 1 1.00000 0.00000 -0.00000 0.00000 0.00000 -1.00000 -0.00000 0.00000 -0.00000 0.00000 -1.00000 0.11597 ******************************************************************************* PAMOC RV 2014-03-01 PAMOC Information on the Molecular System ----------------------------------- mario.barzaghi@cnr.it https://www.pamoc.it ******************************************************************************* Center labels: O1 H2 H3 Symmetry Constraints: H2 -------------------------------------------------------------------------------- IDF reference system with origin at x=0, y=0, z=0 -------------------------------------------------------------------------------- Center Center Atomic Coordinates (Angstroms) Atomic Net Charge Number Label Number X Y Z Mlk Stn EEM -------------------------------------------------------------------------------- 1 O1 8 0.000000 0.000000 0.000000 -0.536 -0.521 -0.410 2 H2 1 0.000000 -0.755684 0.579857 0.261 0.254 0.205 3 H3 1 0.000000 0.755684 0.579857 0.262 0.254 0.205 -------------------------------------------------------------------------------- Center of Mass 0.000000 0.000000 0.064895 Center of Charges 0.000000 0.000000 0.115971 Molecular Charge -0.013 -0.013 0.000 Total number of electrons 10.013 10.013 10.000 Multiplicity 1 Point Group C2V Stoichiometry H2O(0) Principal Moments of Inertia: 0.6019 1.1511 1.7529 (amu-Angstrom**2) Anisotropy: 0.9972 (amu-Angstrom**2) Asphericity (relative anisotropy): 0.2844 Largest atomic distance from the center of mass: 0.9145 (Angstrom) --------------------------------------------------------- Translated and Rotated Cartesion Coordinates (Angstroms) --------------------------------------------------------- Origin: center of mass Reference System: principal axes of the inertial tensor --------------------------------------------------------- Center Center Atomic Coordinates (Angstroms) Number Label Number X Y Z --------------------------------------------------------- 1 O1 8 0.000000 -0.064895 0.000000 2 H2 1 0.755684 0.514962 -0.000000 3 H3 1 -0.755684 0.514962 -0.000000 --------------------------------------------------------- Center of Mass 0.000000 0.000000 0.000000 Center of Charges 0.000000 0.051077 -0.000000 Euler angles (degree): Alpha = 0.000, Beta = 90.000, Gamma = 270.000 | -0.000 -1.000 0.000| Rotation Matrix = | -0.000 0.000 1.000| | -1.000 -0.000 0.000| ******************************************************************************* PAMOC RV 2014-03-01 PAMOC Molecular Electrostatic Multipole Moments (unabridged cartesian tensors) obtained from different DMA of the electron density Standard units: electrons, Debye, Debye-Ang, Debye-Ang**2, Debye-Ang**3 Origin: Center of Mass - Reference System: principal axes of the inertial ten mario.barzaghi@cnr.it https://www.pamoc.it ******************************************************************************* ------------------------------------------------------------- L <O> Mulliken Stone EEM ------------------------------------------------------------- 1 0 0 <q> -0.0128 -0.0128 0.0000 2 1 1 <x> 0.0010 0.0010 0.0000 3 1 2 <y> 1.9880 1.9880 1.1406 4 1 3 <z> -0.0000 -0.0000 -0.0000 Dipole 1.9880 1.9880 1.1406 5 2 1 <xx> -4.1200 -4.1200 1.1233 6 2 2 <xy> -0.0027 -0.0027 0.0000 7 2 3 <yy> -5.7842 -5.7842 0.5133 8 2 4 <xz> 0.0082 0.0082 -0.0000 9 2 5 <yz> -0.0000 -0.0000 -0.0000 10 2 6 <zz> -7.1655 -7.1655 0.0000 (<xx> + <yy> + <zz>)/3 -5.6899 -5.6899 0.5455 Anisotropy 1.6252 1.6252 0.9869 11 3 1 <xxx> 0.0001 0.0001 0.0000 12 3 2 <yyy> 0.2439 0.2439 0.2692 13 3 3 <zzz> 0.0000 0.0000 -0.0000 14 3 4 <xyy> 0.0004 0.0004 0.0000 15 3 5 <xxy> 1.1607 1.1607 0.5784 16 3 6 <xxz> -0.0000 -0.0000 -0.0000 17 3 7 <xzz> 0.0002 0.0002 0.0000 18 3 8 <yzz> -0.1183 -0.1183 0.0000 19 3 9 <yyz> 0.0000 0.0000 -0.0000 20 3 10 <xyz> -0.0001 -0.0001 -0.0000 <xxx> + <xyy> + <xzz> 0.0007 0.0007 0.0000 <yyy> + <xxy> + <yzz> 1.2863 1.2863 0.8476 <zzz> + <xxz> + <yyz> 0.0000 0.0000 -0.0000 Magnitude of vector part 1.2863 1.2863 0.8476 Project. on dipole dir. 1.2863 1.2863 0.8476 21 4 1 <xxxx> -5.4724 -5.4724 0.6415 22 4 2 <yyyy> -5.9036 -5.9036 0.1383 23 4 3 <zzzz> -5.1164 -5.1164 0.0000 24 4 4 <xxxy> -0.0005 -0.0005 0.0000 25 4 5 <xxxz> 0.0033 0.0033 -0.0000 26 4 6 <yyyx> -0.0006 -0.0006 0.0000 27 4 7 <yyyz> -0.0000 -0.0000 -0.0000 28 4 8 <zzzx> 0.0017 0.0017 -0.0000 29 4 9 <zzzy> -0.0000 -0.0000 -0.0000 30 4 10 <xxyy> -1.4367 -1.4367 0.2979 31 4 11 <xxzz> -2.0901 -2.0901 0.0000 32 4 12 <yyzz> -1.9107 -1.9107 0.0000 33 4 13 <xxyz> -0.0000 -0.0000 -0.0000 34 4 14 <yyxz> 0.0003 0.0003 -0.0000 35 4 15 <zzxy> -0.0007 -0.0007 0.0000 <xxxx> + <xxyy> + <xxzz> -8.9992 -8.9992 0.9393 <yyyy> + <xxyy> + <yyzz> -9.2509 -9.2509 0.4362 <zzzz> + <yyzz> + <xxzz> -9.1172 -9.1172 0.0000 <xxxy> + <yyyx> + <zzxy> -0.0017 -0.0017 0.0000 <xxxz> + <zzzx> + <yyxz> 0.0053 0.0053 -0.0000 <yyyz> + <zzzy> + <xxyz> -0.0000 -0.0000 -0.0000 Isotropic value -9.1224 -9.1224 0.4585 ------------------------------------------------------------- ******************************************************************************* PAMOC RV 2014-03-01 PAMOC Molecular Electrostatic Multipole Moments (traceless cartesian tensors) obtained from different DMA of the electron density Standard units: electrons, Debye, Debye-Ang, Debye-Ang**2, Debye-Ang**3 Origin: Center of Mass - Reference System: principal axes of the inertial ten mario.barzaghi@cnr.it https://www.pamoc.it ******************************************************************************* ------------------------------------------------------------- L <O> Mulliken Stone EEM ------------------------------------------------------------- 1 0 0 <q> -0.0128 -0.0128 0.0000 2 1 1 <x> 0.0010 0.0010 0.0000 3 1 2 <y> 1.9880 1.9880 1.1406 4 1 3 <z> -0.0000 -0.0000 -0.0000 Dipole 1.9880 1.9880 1.1406 5 2 1 <xx> 2.3548 2.3548 0.8666 6 2 2 <xy> -0.0041 -0.0041 0.0000 7 2 3 <yy> -0.1415 -0.1415 -0.0483 8 2 4 <xz> 0.0123 0.0123 -0.0000 9 2 5 <yz> -0.0000 -0.0000 -0.0000 10 2 6 <zz> -2.2134 -2.2134 -0.8183 Anisotropy 1.9905 1.9905 1.2087 11 3 1 <xxx> -0.0008 -0.0008 0.0000 12 3 2 <yyy> -1.3196 -1.3196 -0.5985 13 3 3 <zzz> -0.0000 -0.0000 0.0000 14 3 4 <xyy> 0.0006 0.0006 0.0000 15 3 5 <xxy> 2.2585 2.2585 1.0223 16 3 6 <xxz> -0.0000 -0.0000 -0.0000 17 3 7 <xzz> 0.0002 0.0002 -0.0000 18 3 8 <yzz> -0.9390 -0.9390 -0.4238 19 3 9 <yyz> 0.0000 0.0000 -0.0000 20 3 10 <xyz> -0.0002 -0.0002 -0.0000 21 4 1 <xxxx> -0.4575 -0.4575 -0.2003 22 4 2 <yyyy> -1.4000 -1.4000 -0.5148 23 4 3 <zzzz> 1.5425 1.5425 0.5158 24 4 4 <xxxy> 0.0012 0.0012 0.0000 25 4 5 <xxxz> 0.0044 0.0044 -0.0000 26 4 6 <yyyx> 0.0007 0.0007 -0.0000 27 4 7 <yyyz> 0.0000 0.0000 0.0000 28 4 8 <zzzx> -0.0026 -0.0026 0.0000 29 4 9 <zzzy> 0.0000 0.0000 0.0000 30 4 10 <xxyy> 1.7000 1.7000 0.6155 31 4 11 <xxzz> -1.2425 -1.2425 -0.4151 32 4 12 <yyzz> -0.3000 -0.3000 -0.1007 33 4 13 <xxyz> -0.0000 -0.0000 -0.0000 34 4 14 <yyxz> -0.0018 -0.0018 -0.0000 35 4 15 <zzxy> -0.0018 -0.0018 -0.0000 ------------------------------------------------------------- ******************************************************************************* PAMOC RV 2014-03-01 PAMOC Molecular Electrostatic Multipole Moments (spherical tensors) obtained from different DMA of the electron density Standard units: electrons, Debye, Debye-Ang, Debye-Ang**2, Debye-Ang**3 Origin: Center of Mass - Reference System: principal axes of the inertial ten mario.barzaghi@cnr.it https://www.pamoc.it ******************************************************************************* -------------------------------------------- <L, M> Mulliken Stone EEM -------------------------------------------- <0, 0> -0.0128 -0.0128 0.0000 <1,+1> 0.0010 0.0010 0.0000 <1,-1> 1.9880 1.9880 1.1406 <1, 0> -0.0000 -0.0000 -0.0000 <2, 2> 4.9926 4.9926 1.8298 <2,-2> -0.0162 -0.0162 0.0000 <2, 1> 0.0247 0.0247 -0.0000 <2,-1> -0.0000 -0.0000 -0.0000 <2, 0> -2.2134 -2.2134 -0.8183 <3, 3> -0.0157 -0.0157 -0.0000 <3,-3> 48.5708 48.5708 21.9927 <3, 2> -0.0000 -0.0000 -0.0000 <3,-2> -0.0026 -0.0026 -0.0000 <3, 1> 0.0007 0.0007 -0.0000 <3,-1> -2.8169 -2.8169 -1.2714 <3, 0> -0.0000 -0.0000 0.0000 <4, 4> -289.3733 -289.3733 -105.7888 <4,-4> 0.0443 0.0443 0.0000 <4, 3> 0.2360 0.2360 0.0000 <4,-3> -0.0001 -0.0001 -0.0000 <4, 2> -11.3098 -11.3098 -3.7737 <4,-2> -0.0441 -0.0441 -0.0000 <4, 1> -0.0103 -0.0103 0.0000 <4,-1> 0.0000 0.0000 0.0000 <4, 0> 1.5425 1.5425 0.5158 -------------------------------------------- ******************************************************************************* PAMOC RV 2014-03-01 PAMOC Nuclear Centred Distributed Multipoles Analysis - Unabridged cartesian tensors Partitioning schemes: Mulliken Stone Origin: nuclear centres - Reference System: IDF axes Standard units: electrons, Debye, Debye-Ang, Debye-Ang**2, Debye-Ang**3 mario.barzaghi@cnr.it https://www.pamoc.it ******************************************************************************* ---------------------------------------------------- Center L <O> Mulliken Stone ---------------------------------------------------- 8 O1 1 0 0 <q> -0.5359 -0.5207 2 1 1 <x> 0.0000 0.0000 3 1 2 <y> -0.0030 -0.0011 4 1 3 <z> 0.0348 0.0416 5 2 1 <xx> -6.0673 -5.9255 6 2 2 <xy> 0.0068 0.0080 7 2 3 <yy> -5.7219 -5.4920 8 2 4 <xz> 0.0000 0.0000 9 2 5 <yz> 0.0025 0.0026 10 2 6 <zz> -5.9817 -5.7397 11 3 1 <xxx> 0.0000 -0.0000 12 3 2 <yyy> -0.0006 -0.0001 13 3 3 <zzz> -0.1434 -0.0385 14 3 4 <xyy> 0.0000 0.0000 15 3 5 <xxy> -0.0007 -0.0003 16 3 6 <xxz> 0.0115 0.0216 17 3 7 <xzz> 0.0000 -0.0000 18 3 8 <yzz> -0.0002 -0.0000 19 3 9 <yyz> -0.3245 -0.2157 20 3 10 <xyz> 0.0002 0.0004 21 4 1 <xxxx> -4.3813 -4.2129 22 4 2 <yyyy> -3.9951 -3.8613 23 4 3 <zzzz> -4.2863 -3.9981 24 4 4 <xxxy> 0.0014 0.0017 25 4 5 <xxxz> 0.0000 0.0000 26 4 6 <yyyx> 0.0026 0.0032 27 4 7 <yyyz> 0.0004 0.0005 28 4 8 <zzzx> 0.0000 0.0000 29 4 9 <zzzy> 0.0005 0.0005 30 4 10 <xxyy> -1.3724 -1.3296 31 4 11 <xxzz> -1.4363 -1.3672 32 4 12 <yyzz> -1.4688 -1.3571 33 4 13 <xxyz> 0.0006 0.0006 34 4 14 <yyxz> 0.0000 0.0000 35 4 15 <zzxy> 0.0002 0.0003 ---------------------------------------------------- Center L <O> Mulliken Stone ---------------------------------------------------- 1 H2 1 0 0 <q> 0.2612 0.2539 2 1 1 <x> -0.0021 -0.0002 3 1 2 <y> -0.2277 -0.3697 4 1 3 <z> 0.2469 0.2639 5 2 1 <xx> -0.5494 -0.6200 6 2 2 <xy> -0.0008 -0.0000 7 2 3 <yy> -0.2602 -0.5693 8 2 4 <xz> 0.0010 0.0001 9 2 5 <yz> -0.0917 0.0147 10 2 6 <zz> -0.4807 -0.6096 11 3 1 <xxx> -0.0005 -0.0000 12 3 2 <yyy> 0.3569 -0.1576 13 3 3 <zzz> -0.0143 0.1471 14 3 4 <xyy> -0.0007 -0.0000 15 3 5 <xxy> -0.0010 -0.0788 16 3 6 <xxz> 0.0211 0.0570 17 3 7 <xzz> -0.0006 -0.0000 18 3 8 <yzz> 0.0971 -0.0636 19 3 9 <yyz> -0.1349 0.0281 20 3 10 <xyz> 0.0004 0.0000 21 4 1 <xxxx> -0.3676 -0.4517 22 4 2 <yyyy> 0.4292 -0.3671 23 4 3 <zzzz> -0.1805 -0.4482 24 4 4 <xxxy> -0.0002 -0.0000 25 4 5 <xxxz> 0.0002 0.0000 26 4 6 <yyyx> -0.0007 -0.0000 27 4 7 <yyyz> -0.2341 -0.0034 28 4 8 <zzzx> 0.0004 0.0000 29 4 9 <zzzy> -0.1525 0.0112 30 4 10 <xxyy> -0.0467 -0.1453 31 4 11 <xxzz> -0.0998 -0.1522 32 4 12 <yyzz> 0.0671 -0.1336 33 4 13 <xxyz> -0.0291 0.0075 34 4 14 <yyxz> 0.0004 0.0000 35 4 15 <zzxy> -0.0003 -0.0000 ---------------------------------------------------- Center L <O> Mulliken Stone ---------------------------------------------------- 1 H3 1 0 0 <q> 0.2618 0.2540 2 1 1 <x> 0.0021 0.0002 3 1 2 <y> 0.2278 0.3696 4 1 3 <z> 0.2455 0.2638 5 2 1 <xx> -0.5488 -0.6200 6 2 2 <xy> -0.0008 -0.0000 7 2 3 <yy> -0.2611 -0.5693 8 2 4 <xz> -0.0010 -0.0001 9 2 5 <yz> 0.0917 -0.0146 10 2 6 <zz> -0.4798 -0.6095 11 3 1 <xxx> 0.0005 0.0000 12 3 2 <yyy> -0.3555 0.1575 13 3 3 <zzz> -0.0150 0.1471 14 3 4 <xyy> 0.0007 0.0000 15 3 5 <xxy> 0.0010 0.0788 16 3 6 <xxz> 0.0208 0.0570 17 3 7 <xzz> 0.0006 0.0000 18 3 8 <yzz> -0.0972 0.0636 19 3 9 <yyz> -0.1346 0.0281 20 3 10 <xyz> 0.0004 0.0000 21 4 1 <xxxx> -0.3674 -0.4517 22 4 2 <yyyy> 0.4273 -0.3671 23 4 3 <zzzz> -0.1799 -0.4482 24 4 4 <xxxy> -0.0002 -0.0000 25 4 5 <xxxz> -0.0002 -0.0000 26 4 6 <yyyx> -0.0007 -0.0000 27 4 7 <yyyz> 0.2335 0.0034 28 4 8 <zzzx> -0.0004 -0.0000 29 4 9 <zzzy> 0.1525 -0.0112 30 4 10 <xxyy> -0.0470 -0.1453 31 4 11 <xxzz> -0.0996 -0.1522 32 4 12 <yyzz> 0.0668 -0.1336 33 4 13 <xxyz> 0.0291 -0.0074 34 4 14 <yyxz> -0.0004 -0.0000 35 4 15 <zzxy> -0.0003 -0.0000 Electrostatic Properties cpu time: 0.0 seconds.
PAMoC-TUI isn't a visualization program, so that it doesn't provide any
graphical output directly. Its results are printed in tabular form on the
output file and saved on a CUBE file.[L1CUBE
File Format
(PAMoC Manual)] Eventually, the TUI can launch a
suitable visualization program which is able to read the CUBE file.[L1CUBE File Format
(PAMoC
Manual)]
References
Links
- “PAMoC User's Manual: CUBE File Format”. Online
resource:
https://www.pamoc.it/tpc_cube_file_format.html.
Accessed ?, 2018.
